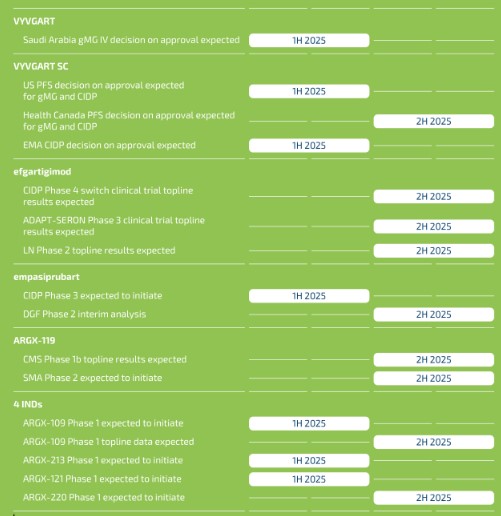
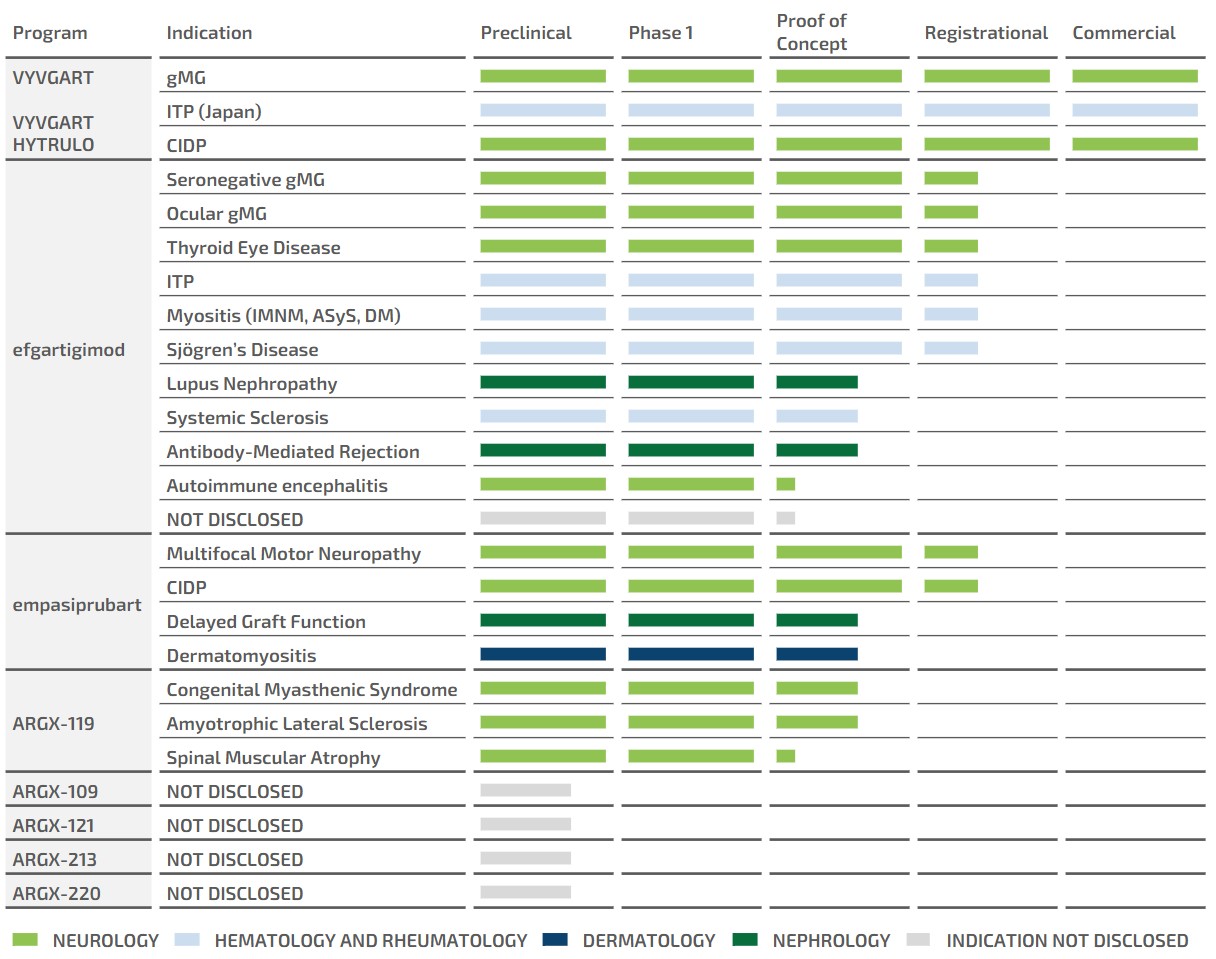
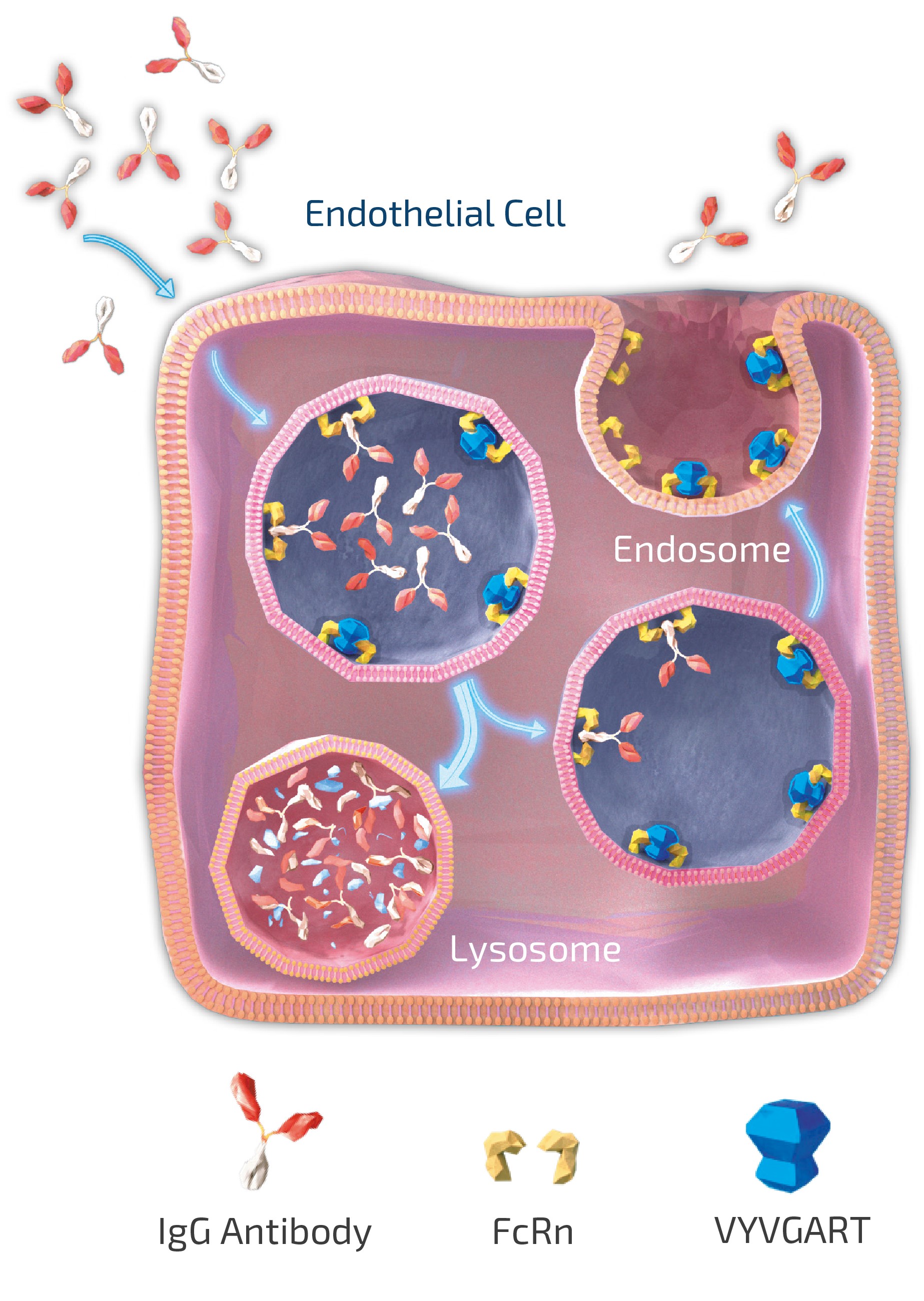
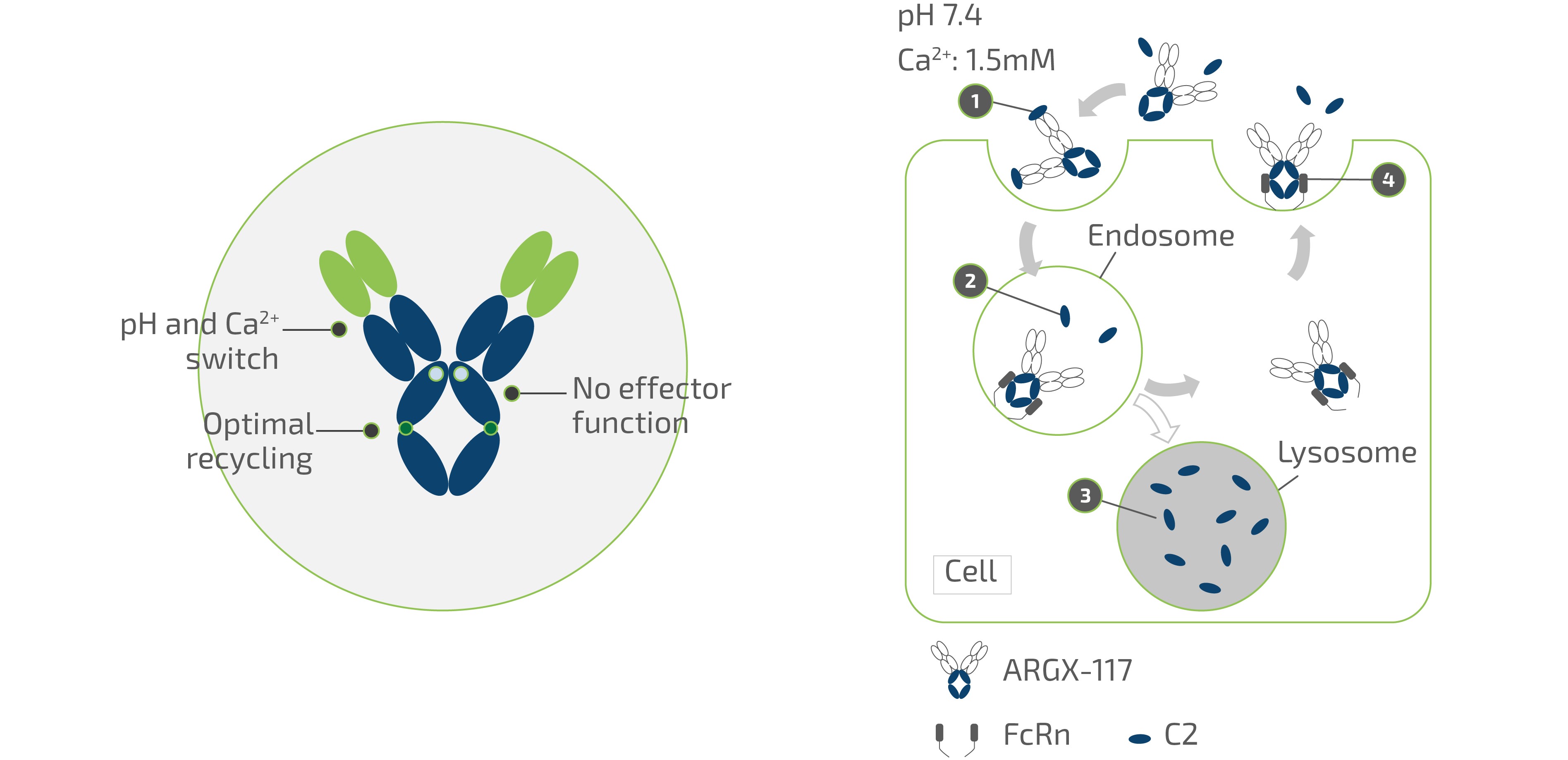
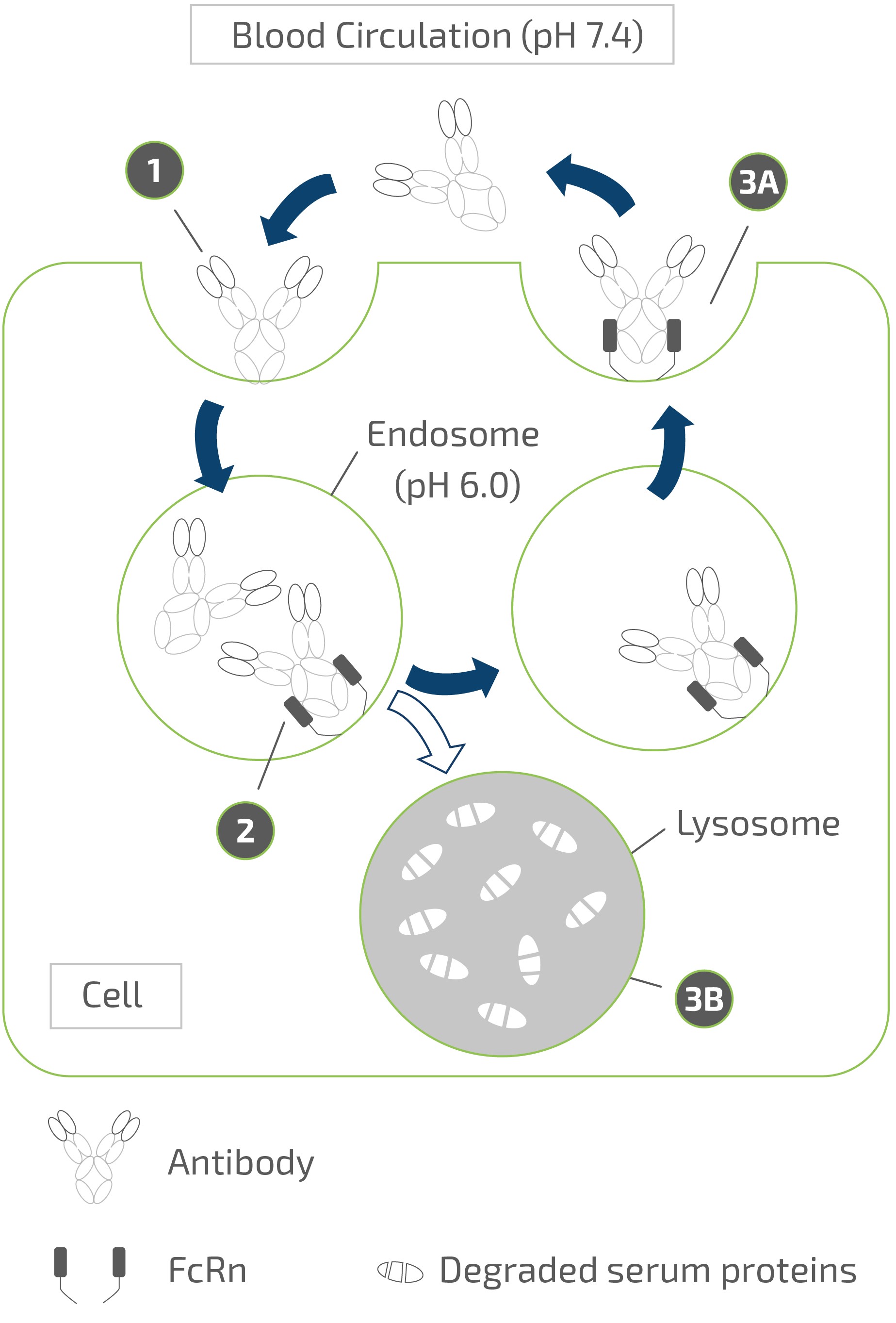
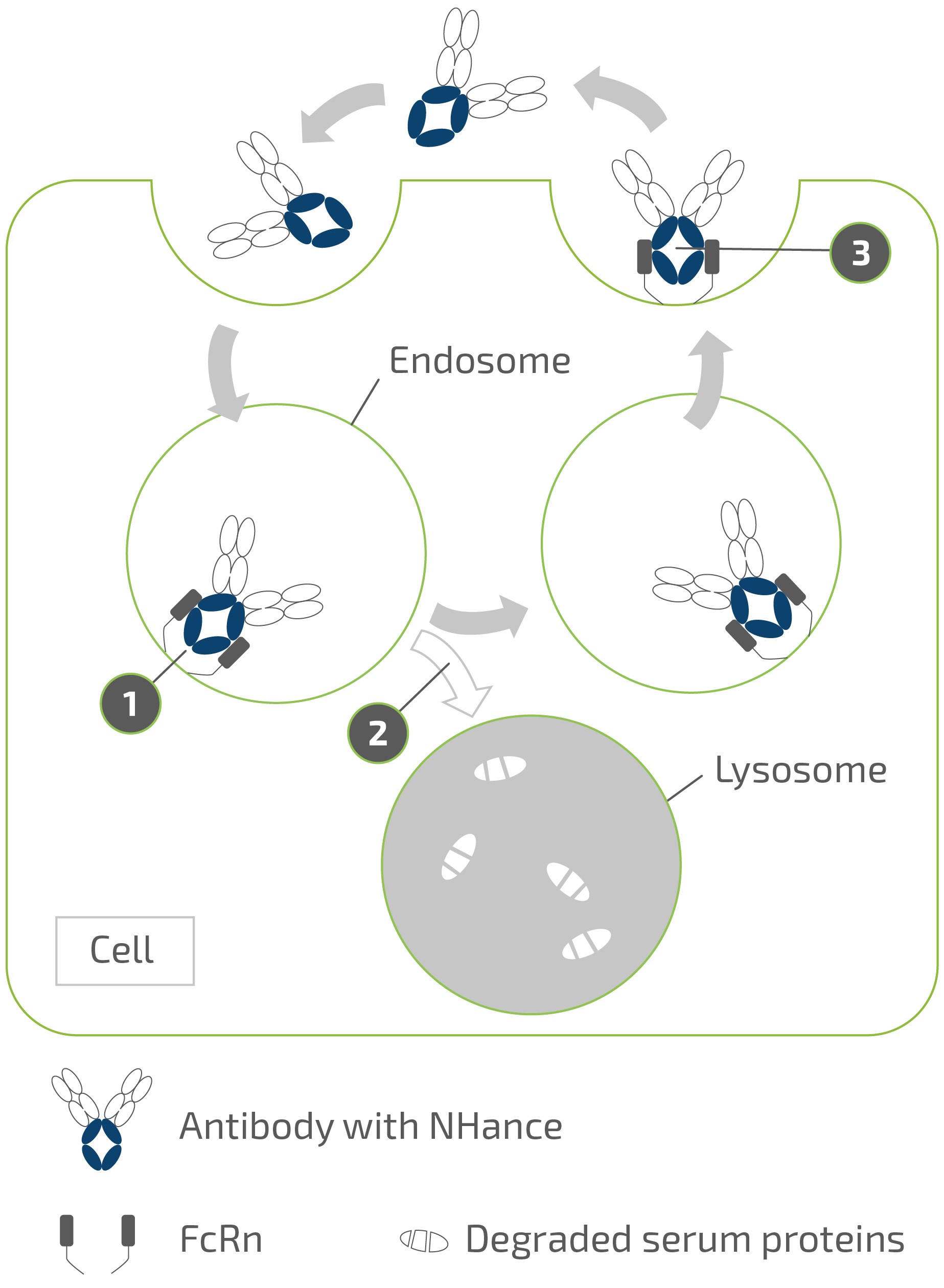
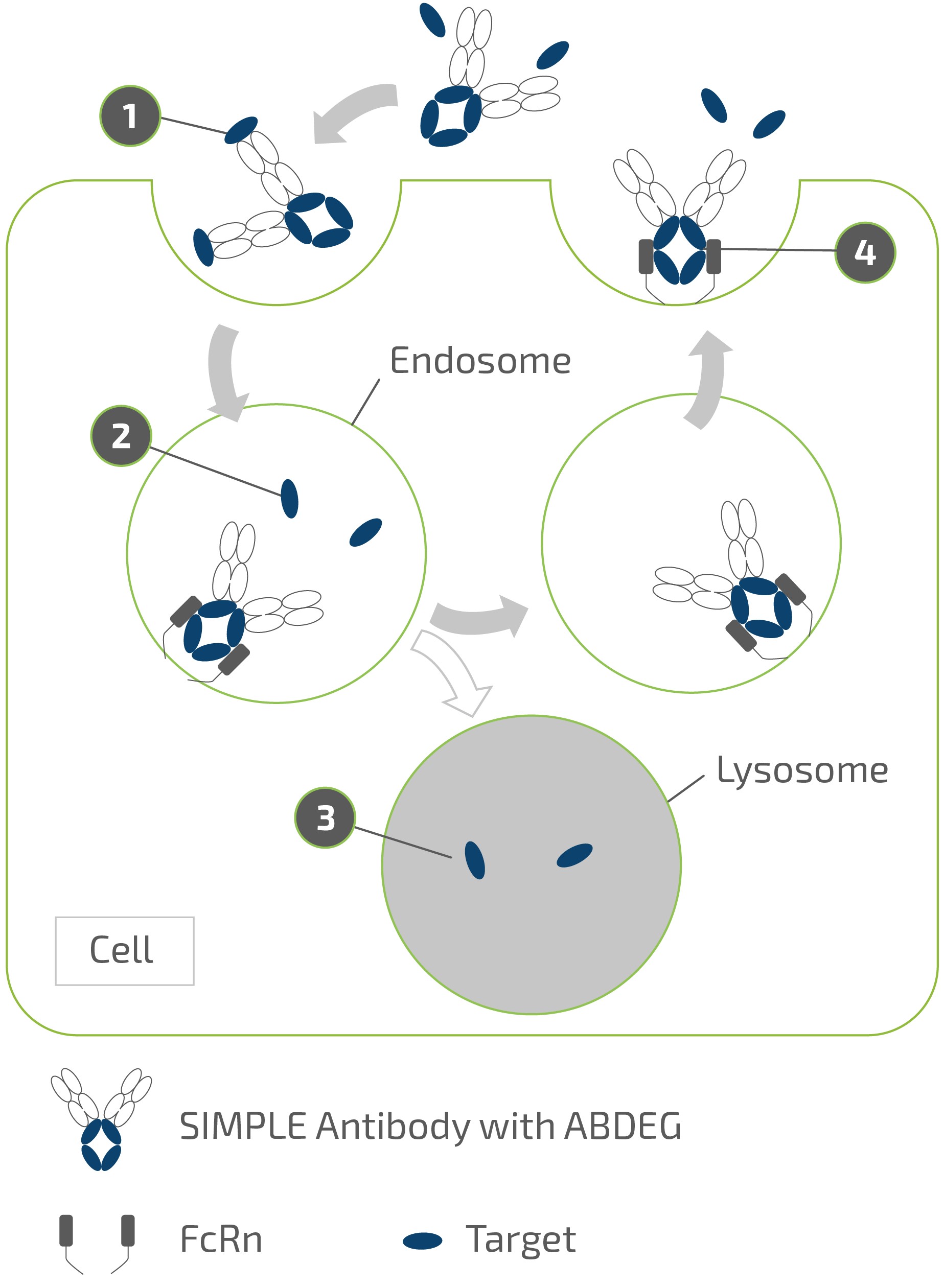
claims challenging the legality of their patient assistance programs under a variety of federal and state laws. It is possible
that we may make grants to independent charitable foundations that help financially needy patients with their premium,
co-pay, and co-insurance obligations. If we choose to do so, and if we or our vendors or donation recipients are deemed
to fail to comply with relevant laws, regulations or evolving government guidance in the operation of these programs, we
could be subject to damages, fines, penalties, or other criminal, civil, or administrative sanctions or enforcement actions.
We cannot ensure that our compliance controls, policies, and procedures will be sufficient to protect against acts of our
employees, business partners, or vendors that may violate the laws or regulations of the jurisdictions in which we
operate. Regardless of whether we have complied with the law, a government investigation could impact our business
practices, harm our reputation, divert the attention of management, increase our expenses, and reduce the availability of
foundation support for our patients who need assistance.
Violations of these laws or any future enacted laws can subject us to criminal, civil and administrative sanctions
including monetary penalties, damages, fines, disgorgement, individual imprisonment and exclusion from participation
in government funded healthcare programs, such as Medicare and Medicaid, additional reporting requirements and
oversight if we become subject to a corporate integrity agreement or similar agreement to resolve allegations of non-
compliance with these laws, reputational harm, and we may be required to curtail or restructure our operations.
Moreover, we expect that there will continue to be federal and state laws and regulations, proposed and implemented,
that could impact our future operations and business.
Because of the breadth of these laws and the narrowness of the statutory exceptions and safe harbors available, it is
possible that some of our business activities could be subject to challenge under one or more of such laws. Ensuring that
our internal operations and future business arrangements with third parties comply with applicable healthcare laws and
regulations will involve substantial costs. It is possible that governmental authorities will conclude that our business
practices do not comply with current or future statutes, regulations, agency guidance or case law involving applicable
fraud and abuse or other healthcare laws and regulations. If our operations are found to be in violation of any of the laws
described above or any other governmental laws and regulations that may apply to us, we may be subject to significant
penalties, including administrative, civil and criminal penalties, damages, fines, disgorgement, the exclusion from
participation in federal and state healthcare programs, individual imprisonment, reputational harm, and the curtailment or
restructuring of our operations, as well as additional reporting obligations and oversight if we become subject to a
corporate integrity agreement or other agreement to resolve allegations of non-compliance with these laws. Further,
defending against any such actions can be costly and time-consuming, and may require significant financial and
personnel resources. Therefore, even if we are successful in defending against any such actions that may be brought
against us, our business may be impaired. If any of the physicians or other providers or entities with whom we expect to
do business are found to not be in compliance with applicable laws, they may be subject to criminal, civil or
administrative sanctions, including exclusions from government funded healthcare programs and imprisonment. If any of
the above occur, our ability to operate our business and our results of operations could be adversely affected.
Healthcare Reform
In the U.S., the EU and other foreign jurisdictions, there have been a number of legislative and regulatory changes to the
healthcare systems that could affect our future results of operations. In particular, there have been and continue to be a
number of initiatives at the U.S. federal and state levels that seek to reduce healthcare costs and improve the quality of
healthcare. For example, the ACA, effective since March 2010, is a sweeping law intended to broaden access to health
insurance, reduce or constrain the growth of healthcare spending, enhance remedies against fraud and abuse, add new
transparency requirements for the healthcare and health insurance industries, impose new taxes and fees on the health
industry and impose additional health policy reforms.
Healthcare reforms that have been adopted, and that may be adopted in the future, could result in further reductions in
coverage and levels of reimbursement for pharmaceutical products, increases in rebates payable under U.S. government
rebate programs and additional downward pressure on pharmaceutical product prices. As discussed above, in August
2022, the IRA was enacted codifying, among other things: a Medicare drug price negotiation program, under which HHS
directly negotiates the selling price of statutorily specified number of Part B and Part D drugs and biologics each year;
inflation rebates which penalizes drug manufacturers that increase prices of Medicare Part B and Part D drugs at a rate
greater than the rate of inflation; and a redesign of the Part D benefit. The IRA permits the Secretary of HHS to
implement many of these provisions through guidance, as opposed to regulation, for the initial years. Manufacturers that
fail to comply with the IRA may be subject to various penalties, including civil monetary penalties. The IRA also
extends enhanced subsidies for individuals purchasing health insurance coverage in ACA marketplaces through plan
year 2025. These provisions will take began taking progressively starting in 2023, although certain policies have been
subject to legal challenges. For example, the provisions related to the negotiation of selling prices of high-expenditure
single-source drugs and biologics have been challenged in multiple lawsuits. Additionally, we cannot predict whether the
U.S. Congress will amend the IRA or if the government will adopt new or different interpretations of the law in future
guidance or rulemaking. However, at this time, the Trump administration is continuing to implement the IRA and to
defend the law in litigation. While it is unclear how the IRA will be implemented in the future and the outcome of the
litigation, it will likely have a significant impact on the pharmaceutical industry.